What is the diagnostics market?
Distinctions in diagnostics are made between in vitro tests, imaging agents and diagnostic devices. In vitro diagnostics analyse samples taken from the body, including hair or blood. Pregnancy and rapid HIV tests, along with DNA sequencing machines are often regulated as devices.
Diagnostic devices, whether mechanical, optical or electric, can range from the oldest and simplest tools used to help identify disease, eg a stethoscope, to complex MRI machines.
Finally, imaging agents are substances injected into a patient that help in the identification of structures and substances in the body using radiographic equipment. Both MRI contrast agents and PET tracers in the detection of beta amyloid for the diagnosis of Alzheimer’s disease are examples of imaging agents.
How is the success or failure of a diagnostic test evaluated?
One of the most commonly used measures of a diagnostic test’s efficacy is the percentage of true positive to true negative results it returns. In a perfect case study, you would expect a test where all patients who are positive (P) or negative (N) for a condition are represented by an accurate true positive (TP) or true negative (TN) result. By evaluating TP rates, we can define a test’s ‘sensitivity’, or true positive rate (TP/P).
TP/P, the proportion of positive results that are truly accurate, is directly related to the false negative rate (FN/P), or the ratio of those patients who had a disease but received a false negative result.
If a test has a sensitivity rating of 99%, it would therefore have an FN/P false positive rate of 1%. The other side of sensitivity is a test’s specificity, also known as its true negative rate. The true negative rate (TN/N) is naturally related to a test’s false positive ratio, in a similar way to sensitivity. A specificity rate of 90% therefore has a false positive rate of 10%.
How are sensitivity and specificity interrelated?
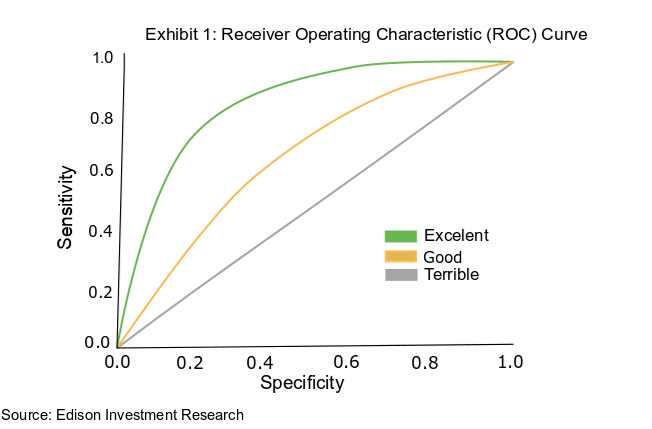
There is a balance to be struck between a test’s specificity and sensitivity. In some cases, this is due to the threshold at which a test returns a positive result. For example, a test that reads biomarkers in the blood will set a certain concentration of markers that represent a positive result.
If that threshold is too high, the test would deliver a very high proportion of true positive results, given that only those patients with severe cases of a disease would produce sufficient biomarkers to pass the test’s threshold. Such a test would have a high level of sensitivity, but by extension a low specificity.
This is because those who suffer only mildly from the disease or are in its earliest stages have fewer biomarkers in their blood than the high threshold set for the test, will receive a false negative result initially and may go on to test positive. The relationship between specificity and sensitivity is captured in a receiver operating characteristic (ROC) curve (Exhibit 1).
How do we discover a test’s efficacy in clinical trials?
Clinical studies in the diagnostics field are not well defined in the same way as Phase I, II and III studies in the therapeutics field. The level of clinical data needed for approval in a diagnostic study can vary significantly, depending on the use and design of the test.
In addition, these studies may need to test hundreds or even thousands of patient samples prospectively so may not be as expensive as a drug trial, but can take considerable time as the experimental diagnostic results are correlated with either a gold standard test, or a clinical diagnosis. However, even with their diversity, clinical studies can still be broadly split into retrospective and prospective studies.
A retrospective study involves collecting samples from patients previously tested for or diagnosed with a disease and comparing the results of the test with medical records. Retrospective studies are regularly conducted during the early stages of a test’s development, since they are cheap and useful for working out the basic parameters/guidelines of a newly developed test.
Later in development, prospective studies are the norm required for certain regulatory approvals. Here, the parameters of a test are decided before the trial, prospectively. The tests are then carried out in the setting in which the drug will be used on the market, as patients present at a clinic. The patients are then tested with established methods and the results compared. Prospective studies are nearly always used when seeking approval for a new diagnostic, as the results gathered more closely reflect real world conditions.
How else do real world conditions relate to diagnostic studies?
Developing a highly accurate test is not necessarily enough to gain regulatory approval. Developers also have to prove that their test is clinically useful, can affect patient outcomes and, in the case of point-of-care (PoC) diagnostics, is robust enough to be used outside a laboratory environment, and even by the patient at home.
There have been many cases where tests have been clinically accurate but failed to prove clinically valid. For example, the prostate-specific antigen (PSA) test for prostate cancer has a high degree of accuracy, with a specificity of 85%. However, the test is not recommended for those over 70 years of age by a number of national bodies in their clinical guidelines, including the US Preventive Services Task Force. This is because PSA screening has been found, in some clinical utility studies, to have no impact on mortality rates in those over 70, given the long course of the disease.
Companies do not always have to demonstrate clinical utility through a study to get approval for a product. That said, utility studies are often used by regulatory bodies when determining treatment guidelines, and can be important indicators of future commercial success.


